
Nuvigil: Tratamiento de somnolencia excesiva (información de prescripción completa)
Nombre de la marca: Nuvigil
Nombre genérico: armodafinilo
Tabletas de Nuvigil® (armodafinilo) [C-IV]
El armodafinilo es un medicamento que promueve la vigilia disponible cuando Nuvigil se usa para tratar la apnea del sueño, la narcolepsia o el trastorno del sueño en el trabajo por turnos. Uso, dosificación, efectos secundarios.
Contenido:
Descripción
Farmacología Clínica
Ensayos clínicos
Indicaciones y uso
Contraindicaciones
Advertencias
Precauciones
Reacciones adversas
Abuso y dependencia de drogas
Sobredosis
Dosificación y administración
Cómo suministrado
Nuvigil hoja de información para el paciente (en inglés simple)
Descripción
NUVIGIL® (armodafinilo) es un agente promotor de la vigilia para la administración oral. El armodafinilo es el enantiómero R del modafinilo que es una mezcla de los enantiómeros R y S. El nombre químico del armodafinilo es 2 - [(R) - (difenilmetil) sulfinil] acetamida. La fórmula molecular es C15H15NO2S y el peso molecular es 273.35.
La estructura química es:
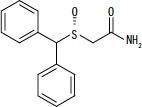
El armodafinilo es un polvo cristalino blanco a blanquecino que es muy poco soluble en agua, poco soluble en acetona y soluble en metanol. Las tabletas de NUVIGIL contienen 50, 150 o 250 mg de armodafinilo y los siguientes ingredientes inactivos: croscarmelosa sodio, lactosa monohidrato, estearato de magnesio, celulosa microcristalina, povidona y pregelatinizada almidón.
parte superior
Farmacología Clínica
Mecanismo de acción y farmacología.
Se desconocen los mecanismos precisos a través de los cuales el armodafinilo (enantiómero R) o el modafinilo (mezcla de enantiómeros R y S) promueven la vigilia. Tanto el armodafinilo como el modafinilo han mostrado propiedades farmacológicas similares en estudios no clínicos en animales e in vitro, en la medida de lo probado.
continuar la historia a continuación
A concentraciones farmacológicamente relevantes, el armodafinilo no se une o inhibe a varios receptores y enzimas potencialmente relevantes para regulación del sueño / vigilia, incluidas las de serotonina, dopamina, adenosina, galanina, melatonina, melanocortina, orexina-1, orfanina, PACAP o benzodiacepinas o transportadores de GABA, serotonina, norepinefrina y colina o fosfodiesterasa VI, COMT, transaminasa de GABA y tirosina hidroxilasa El modafinilo no inhibe la actividad de MAO-B o fosfodiesterasas II-IV.
La vigilia inducida por modafinilo puede ser atenuada por el antagonista del receptor adrenérgico α1, la prazosina; sin embargo, el modafinilo está inactivo en otros sistemas de ensayo in vitro que se sabe que responden a agonistas α-adrenérgicos como la preparación deferente de rata.
El armodafinilo no es un agonista del receptor de dopamina de acción directa o indirecta. Sin embargo, in vitro, tanto el armodafinilo como el modafinilo se unen al transportador de dopamina e inhiben la recaptación de dopamina. Para el modafinilo, esta actividad se ha asociado in vivo con un aumento de los niveles extracelulares de dopamina en algunas regiones del cerebro de los animales. En ratones genéticamente modificados que carecían del transportador de dopamina (DAT), el modafinilo carecía de actividad promotora de la estela, lo que sugiere que esta actividad era dependiente de DAT. Sin embargo, los efectos que promueven la estela del modafinilo, a diferencia de los de la anfetamina, no fueron antagonizados por el antagonista del receptor de dopamina haloperidol en ratas.
Además, la alfa-metil-p-tirosina, un inhibidor de la síntesis de dopamina, bloquea la acción de la anfetamina, pero no bloquea la actividad locomotora inducida por el modafinilo.
El armodafinilo y el modafinilo tienen acciones de promoción de la estela similares a los agentes simpaticomiméticos, incluida la anfetamina. y metilfenidato, aunque su perfil farmacológico no es idéntico al del simpaticomimético aminas Además de sus efectos que promueven la estela y su capacidad para aumentar la actividad locomotora en animales, el modafinilo produce Efectos psicoactivos y eufóricos, alteraciones en el estado de ánimo, la percepción, el pensamiento y los sentimientos típicos de otros estimulantes del SNC. Inhumanos. El modafinilo tiene propiedades de refuerzo, como lo demuestra su autoadministración en monos previamente entrenados para autoadministrarse cocaína; el modafinilo también fue parcialmente discriminado como estimulante.
Según estudios no clínicos, dos metabolitos principales, ácido y sulfona, de modafinilo o armodafinilo, no parecen contribuir a las propiedades activadoras del SNC de los compuestos originales.
Farmacocinética
El componente activo de NUVIGIL es el armodafinilo, que es el enantiómero de modafinilo de mayor duración. NUVIGIL exhibe una cinética lineal independiente del tiempo después de la administración de dosis orales únicas y múltiples. El aumento de la exposición sistémica es proporcional en el rango de dosis de 50 a 400 mg. No se observó un cambio dependiente del tiempo en la cinética durante las 12 semanas de dosificación. El estado estacionario aparente para NUVIGIL se alcanzó dentro de los 7 días de la dosificación. En estado estacionario, la exposición sistémica a NUVIGIL es 1,8 veces la exposición observada después de una dosis única. Los perfiles de concentración-tiempo del enantiómero R puro después de la administración de 50 mg de NUVIGIL o 100 mg de PROVIGIL® (modafinilo) son casi superponibles.
Absorción
NUVIGIL se absorbe fácilmente después de la administración oral. La biodisponibilidad oral absoluta no se determinó debido a la insolubilidad acuosa de armodafinilo, que impidió la administración intravenosa. Las concentraciones plasmáticas máximas se alcanzan aproximadamente a las 2 horas en ayunas. El efecto alimentario sobre la biodisponibilidad general de NUVIGIL se considera mínimo; sin embargo, tiempo para alcanzar la concentración máxima (tmax) puede demorarse aproximadamente de 2 a 4 horas en el estado alimentado. Desde el retraso en tmax también se asocia con niveles plasmáticos elevados más adelante en el tiempo, los alimentos pueden afectar potencialmente el inicio y el curso temporal de la acción farmacológica para NUVIGIL.
Distribución
NUVIGIL tiene un volumen aparente de distribución de aproximadamente 42 L. Los datos específicos de la unión a proteínas de armodafinilo no están disponibles. Sin embargo, el modafinilo se une moderadamente a las proteínas plasmáticas (aproximadamente el 60%), principalmente a la albúmina. El potencial de interacciones de NUVIGIL con fármacos altamente unidos a proteínas se considera mínimo.
Metabolismo
Los datos in vitro e in vivo muestran que el armodafinilo sufre desamidación hidrolítica, oxidación S e hidroxilación de anillos aromáticos, con la posterior conjugación de glucurónidos de los productos hidroxilados. La hidrólisis de la amida es la vía metabólica más destacada, y la formación de sulfona por el citocromo P450 (CYP) 3A4 / 5 es la siguiente en importancia. Los otros productos oxidativos se forman muy lentamente in vitro para permitir la identificación de la (s) enzima (s) responsable (s). Solo dos metabolitos alcanzan concentraciones apreciables en plasma (es decir, ácido R-modafinilo y modafinil sulfona).
Los datos específicos de la disposición de NUVIGIL no están disponibles. Sin embargo, el modafinilo se elimina principalmente a través del metabolismo, predominantemente en el hígado, con menos del 10% del compuesto original excretado en la orina. Se recuperó un total de 81% de la radiactividad administrada en 11 días después de la dosis, predominantemente en la orina (80% vs 1.0% en las heces).
Eliminación
Después de la administración oral de NUVIGIL, el armodafinilo exhibe una aparente disminución monoexponencial de la concentración plasmática máxima. La terminal aparente t ½ es de aproximadamente 15 horas. El aclaramiento oral de NUVIGIL es de aproximadamente 33 ml / min.
Interacciones farmacológicas
La existencia de múltiples vías para el metabolismo del armodafinilo, así como el hecho de que una vía no relacionada con el CYP es la más rápida en la metabolización del armodafinilo, sugieren que existe una baja probabilidad de efectos sustantivos en el perfil farmacocinético general de NUVIGIL debido a la inhibición del CYP por concomitante medicamentos
Los datos in vitro demostraron que el armodafinilo muestra una respuesta inductiva débil para CYP1A2 y posiblemente CYP3A actividades de una manera relacionada con la concentración y que la actividad CYP2C19 es reversiblemente inhibida por armodafinilo Otras actividades del CYP no parecieron verse afectadas por el armodafinilo. Un estudio in vitro demostró que el armodafinilo es un sustrato de la glicoproteína P.
La administración crónica de NUVIGIL a 250 mg redujo la exposición sistémica al midazolam en un 32% y 17% después de una sola administración oral (5). mg) e intravenosa (2 mg) dosis, respectivamente, lo que sugiere que la administración de NUVIGIL induce moderadamente CYP3A actividad. Los medicamentos que son sustratos del CYP3A4 / 5, como la ciclosporina, pueden requerir un ajuste de dosis. (Ver Precauciones, Interacciones con la drogas).
La administración crónica de NUVIGIL a 250 mg no afectó la farmacocinética de cafeína (200 mg), un sustrato de sonda para la actividad de CYP1A2.
La administración conjunta de una dosis única de 400 mg de NUVIGIL con omeprazol (40 mg) aumentó sistémicamente exposición al omeprazol en aproximadamente un 40%, lo que indica que el armodafinilo inhibe moderadamente el CYP2C19 actividad. Los medicamentos que son sustratos para CYP2C19 pueden requerir una reducción de la dosis. (Ver Precauciones, Interacciones con la drogas).
Efecto de género
El análisis farmacocinético de la población no sugiere ningún efecto de género en la farmacocinética del armodafinilo.
Poblaciones Especiales
Los datos específicos del armodafinilo en poblaciones especiales no están disponibles.
Efecto de edad: Se observó una ligera disminución (~ 20%) en el aclaramiento oral (CL / F) de modafinilo en un estudio de dosis única a 200 mg en 12 sujetos con una edad media de 63 años (rango 53-72 años), pero no se consideró que el cambio fuera clínicamente significativo. En un estudio de dosis múltiples (300 mg / día) en 12 pacientes con una edad media de 82 años (rango 67-87 años), la media los niveles de modafinilo en plasma fueron aproximadamente dos veces mayores que los obtenidos históricamente en pacientes más jóvenes asignaturas. Debido a los posibles efectos de los múltiples medicamentos concomitantes con los que la mayoría de los pacientes estaban siendo tratada, la diferencia aparente en la farmacocinética de modafinilo puede no ser atribuible únicamente a los efectos de envejecimiento. Sin embargo, los resultados sugieren que el aclaramiento de modafinilo puede reducirse en los ancianos (Ver Dosificación y administración).
Efecto de raza: La influencia de la raza en la farmacocinética de modafinilo no se ha estudiado.
Insuficiencia renal: En un estudio de dosis única de 200 mg de modafinilo, la insuficiencia renal crónica grave (aclaramiento de creatinina â ‰ ¤20 ml / min) no influye significativamente en la farmacocinética de modafinilo, pero la exposición al ácido de modafinilo aumentó 9 veces (ver Precauciones).
Insuficiencia hepática: se examinó la farmacocinética y el metabolismo del modafinilo en pacientes con cirrosis hepática (6 hombres y 3 mujeres). Tres pacientes tenían cirrosis en estadio B o B + y 6 pacientes tenían cirrosis en estadio C o C + (según los criterios de puntuación de Child-Pugh). Clínicamente 8 de 9 pacientes eran ictéricos y todos tenían ascitis. En estos pacientes, el aclaramiento oral de modafinilo se redujo en aproximadamente un 60% y la concentración en estado estacionario se duplicó en comparación con los pacientes normales. La dosis de NUVIGIL debe reducirse en pacientes con insuficiencia hepática grave (ver Precauciones y Dosificación y administración).
parte superior
Ensayos clínicos
La eficacia de NUVIGIL para mejorar la vigilia se ha establecido en el sueño siguiente trastornos: síndrome de apnea obstructiva del sueño / hipopnea (SAHS), narcolepsia y trastorno del sueño en el trabajo por turnos (SWSD)
Para cada ensayo clínico, se requirió un valor p de â ‰ ¤0.05 para la significación estadística.
Síndrome de apnea obstructiva del sueño / hipopnea (SAHS)
La efectividad de NUVIGIL para mejorar la vigilia en pacientes con somnolencia excesiva asociada con SAHS se estableció en dos estudios doble ciego de 12 semanas, multicéntricos, controlados con placebo, de grupos paralelos, de pacientes ambulatorios que se reunieron con el International Criterios de clasificación de los trastornos del sueño (ICSD) para OSAHS (que también son consistentes con la Asociación Americana de Psiquiatría DSM-IV criterios). Estos criterios incluyen: 1) somnolencia excesiva o insomnio, además de episodios frecuentes de discapacidad. respiración durante el sueño y características asociadas como ronquidos fuertes, dolores de cabeza matutinos o sequedad de boca despertar; o 2) somnolencia excesiva o insomnio; y polisomnografía que demuestra uno de los siguientes: más de cinco apneas obstructivas, cada una de más de 10 segundos de duración, por hora de sueño; y uno o más de los siguientes: excitaciones frecuentes del sueño asociadas con las apneas, bradicardia o desaturación arterial de oxígeno en asociación con las apneas. Además, para ingresar a estos estudios, todos los pacientes debían tener somnolencia excesiva como se demostró por un puntaje â ‰ ¥ 10 en la escala de somnolencia de Epworth, a pesar del tratamiento con presión positiva continua en las vías respiratorias (CPAP) Se requirió evidencia de que la CPAP fue efectiva para reducir los episodios de apnea / hipopnea junto con la documentación del uso de la CPAP.
Se requería que los pacientes cumplieran con CPAP, definido como el uso de CPAP â ‰ ¥ 4 horas / noche en â ‰ ¥ 70% de las noches. El uso de CPAP continuó durante todo el estudio. En ambos estudios, las principales medidas de efectividad fueron 1) la latencia del sueño, según lo evaluado por la Prueba de mantenimiento de la vigilia (MWT) y 2) el cambio en el estado general de la enfermedad del paciente, según lo medido por la Impresión Clínica Global de Cambio (CGI-C) al final visitar. Para un ensayo exitoso, ambas medidas tuvieron que mostrar una mejora estadísticamente significativa.
El MWT mide la latencia (en minutos) hasta el inicio del sueño. Se realizó un MWT extendido con sesiones de prueba a intervalos de 2 horas entre las 9 a.m. y las 7 p.m. El análisis primario fue el promedio de las latencias del sueño de las primeras cuatro sesiones de prueba (9AM a 3PM). Para cada sesión de prueba, se le pidió al sujeto que intentara permanecer despierto sin usar medidas extraordinarias. Cada sesión de prueba se terminó después de 30 minutos si no se produjo el sueño o inmediatamente después del inicio del sueño. El CGI-C es una escala de 7 puntos, centrada en Sin cambios, y que va de Mucho peor a Muy mejorada. Los evaluadores no recibieron ninguna orientación específica sobre los criterios que debían aplicar al calificar a los pacientes.
En el primer estudio, un total de 395 pacientes con SAHS se aleatorizaron para recibir NUVIGIL 150 mg / día, NUVIGIL 250 mg / día o un placebo equivalente. Los pacientes tratados con NUVIGIL mostraron una mejora estadísticamente significativa en la capacidad de permanecer despierto en comparación con los pacientes tratados con placebo según lo medido por el MWT en la visita final. Un mayor número estadísticamente significativo de pacientes tratados con NUVIGIL mostró una mejoría en el estado clínico general según la clasificación de la escala CGI-C en la visita final. Las latencias promedio de sueño (en minutos) en el MWT al inicio del estudio se muestran en la Tabla 1 a continuación, junto con el cambio promedio desde el inicio en el MWT en la visita final. Los porcentajes de pacientes que mostraron algún grado de mejora en el CGI-C en los ensayos clínicos se muestran en la Tabla 2 a continuación. Las dos dosis de NUVIGIL produjeron efectos estadísticamente significativos de magnitudes similares en el MWT y también en el CGI-C.
En el segundo estudio, 263 pacientes con SAHS fueron asignados al azar a NUVIGIL 150 mg / día o placebo. Los pacientes tratados con NUVIGIL mostraron una mejora estadísticamente significativa en la capacidad de permanecer despierto en comparación con los pacientes tratados con placebo según lo medido por el MWT [Tabla 1]. Un mayor número estadísticamente significativo de pacientes tratados con NUVIGIL mostró una mejoría en el estado clínico general según la escala de CGI-C [Tabla 2].
El sueño nocturno medido con polisomnografía no se vio afectado por el uso de NUVIGIL en ninguno de los estudios.
Narcolepsia
Se estableció la efectividad de NUVIGIL para mejorar la vigilia en pacientes con somnolencia excesiva (ES) asociada con narcolepsia en un estudio doble ciego de 12 semanas, multicéntrico, controlado con placebo, de grupos paralelos, de pacientes ambulatorios que cumplieron los criterios de ICSD para narcolepsia Un total de 196 pacientes fueron asignados al azar para recibir NUVIGIL 150 o 250 mg / día, o un placebo equivalente. Los criterios de ICSD para la narcolepsia incluyen 1) siestas recurrentes durante el día o lapsos en el sueño que ocurren casi a diario durante al menos tres meses, más pérdida repentina bilateral del tono muscular postural en asociación con emoción intensa (cataplejía), o 2) una queja de exceso somnolencia o debilidad muscular repentina con características asociadas: parálisis del sueño, alucinaciones hipnagógicas, comportamientos automáticos, trastornos mayores episodio de sueño y polisomnografía que demuestra uno de los siguientes: latencia del sueño inferior a 10 minutos o latencia del sueño de movimiento ocular rápido (REM) inferior a 20 minutos y un sueño múltiple Prueba de latencia (MSLT) que demuestra una latencia media del sueño de menos de 5 minutos y dos o más períodos REM de inicio del sueño y ningún trastorno médico o mental explica el síntomas Para ingresar a estos estudios, todos los pacientes debían haber documentado objetivamente la somnolencia diurna excesiva, a través de MSLT con una latencia del sueño de 6 minutos o menos y la ausencia de cualquier otro médico o psiquiátrico activo clínicamente significativo trastorno. El MSLT, una evaluación polisomnográfica objetiva de la capacidad del paciente para conciliar el sueño en un entorno no estimulante, latencia medida (en minutos) para el inicio del sueño en promedio durante 4 sesiones de prueba en Intervalos de 2 horas. Para cada sesión de prueba, se le dijo al sujeto que se acostara en silencio e intentara dormir. Cada sesión de prueba se terminó después de 20 minutos si no se produjo el sueño o inmediatamente después del inicio del sueño.
Las principales medidas de efectividad fueron: 1) latencia del sueño según lo evaluado por la Prueba de mantenimiento de la vigilia (MWT) y 2) el cambio en la enfermedad general del paciente estado, según lo medido por la Impresión clínica global de cambio (CGI-C) en la visita final (consulte la sección PRUEBAS CLÍNICAS, OSAHS anterior para obtener una descripción de estos medidas). Cada sesión de prueba de MWT se terminó después de 20 minutos si no se produjo el sueño o inmediatamente después del inicio del sueño en este estudio.
Los pacientes tratados con NUVIGIL mostraron una capacidad estadísticamente mejorada para permanecer despierto con el MWT en cada dosis en comparación con el placebo en la visita final [Tabla 1]. Un mayor número estadísticamente significativo de pacientes tratados con NUVIGIL en cada dosis mostró una mejoría en el estado clínico general según la clasificación de la escala CGI-C en la visita final [Tabla 2].
Las dos dosis de NUVIGIL produjeron efectos estadísticamente significativos de magnitudes similares en el CGI-C. Aunque se observó un efecto estadísticamente significativo sobre el MWT para cada dosis, se observó que la magnitud del efecto era mayor para la dosis más alta.
El sueño nocturno medido con polisomnografía no se vio afectado por el uso de NUVIGIL.
Trastorno del sueño por turnos de trabajo (SWSD)
La efectividad de NUVIGIL para mejorar la vigilia en pacientes con somnolencia excesiva asociada con SWSD se demostró en un estudio clínico de 12 semanas, multicéntrico, doble ciego, controlado con placebo, de grupos paralelos. juicio. Un total de 254 pacientes con SWSD crónica fueron aleatorizados para recibir NUVIGIL 150 mg / día o placebo. Todos los pacientes cumplieron con los criterios ICSD para SWSD crónico [que son consistentes con los criterios DSM-IV de la Asociación Americana de Psiquiatría para el trastorno del sueño del ritmo circadiano: tipo de trabajo por turnos]. Estos criterios incluyen 1): a) una queja primaria de somnolencia excesiva o insomnio que se asocia temporalmente con un período de trabajo (generalmente trabajo nocturno) que ocurre durante la fase habitual del sueño, o b) la polisomnografía y el MSLT demuestran la pérdida de un patrón normal de sueño-vigilia (es decir, alteración cronobiológica ritmicidad); y 2) ningún otro trastorno médico o mental explica los síntomas, y 3) los síntomas no cumplen con los criterios para cualquier otro trastorno del sueño que produzca insomnio o somnolencia excesiva (por ejemplo, cambio de zona horaria [jet lag] síndrome).
Cabe señalar que no todos los pacientes con una queja de somnolencia que también participan en turnos cumplen con los criterios para el diagnóstico de SWSD. En el ensayo clínico, solo se incluyeron pacientes sintomáticos durante al menos 3 meses.
Los pacientes inscritos también debían trabajar un mínimo de 5 turnos nocturnos por mes, tener somnolencia excesiva en el hora de sus turnos nocturnos (puntuación MSLT â ‰ ¤6 minutos), y tener insomnio durante el día documentado por un polisomnograma diurno (PSG)
Las principales medidas de efectividad fueron 1) latencia del sueño, según lo evaluado por la Prueba de latencia múltiple del sueño (MSLT) realizada durante un turno nocturno simulado a la visita final, y 2) el cambio en el estado general de la enfermedad del paciente, medido por la Impresión clínica global de cambio (CGI-C) en la visita final. (Ver Ensayos clínicos, Narcolepsia y OSAHS secciones anteriores para la descripción de estas medidas).
Los pacientes tratados con NUVIGIL mostraron una prolongación estadísticamente significativa en el tiempo de sueño. inicio comparado con pacientes tratados con placebo, medido por el MSLT nocturno en la visita final [Tabla 1]. Un mayor número estadísticamente significativo de pacientes tratados con NUVIGIL mostró una mejoría en el estado clínico general según la escala de CGI-C en la visita final [Tabla 2].
El sueño diurno medido con polisomnografía no se vio afectado por el uso de NUVIGIL.
| Trastorno | Medida | Nuvigil 150 mg * |
Nuvigil 250 mg * |
Placebo | |||
* Significativamente diferente al placebo para todos los ensayos (p <0.05) | |||||||
| Base | Cambio de la línea de base |
Base | Cambio de la línea de base |
Base | Cambiar de Base |
||
| OSAHS I | MWT | 21.5 | 1.7 | 23.3 | 2.2 | 23.2 | -1.7 |
| OSAHS II | MWT | 23.7 | 2.3 | - | - | 23.3 | -1.3 |
| Narcolepsia | MWT | 12.1 | 1.3 | 9.5 | 2.6 | 12.5 | -1.9 |
| SWSD | MSLT | 2.3 | 3.1 | - | - | 2.4 | 0.4 |
| Trastorno | Nuvigil 150 mg * |
Nuvigil 250 mg * |
Placebo |
* Significativamente diferente al placebo para todos los ensayos (p <0.05) | |||
| OSAHS I | 71% | 74% | 37% |
| OSAHS II | 71% | - | 53% |
| Narcolepsia | 69% | 73% | 33% |
| SWSD | 79% | - | 59% |
parte superior
Indicaciones y uso
NUVIGIL está indicado para mejorar la vigilia en pacientes con somnolencia excesiva asociada con el síndrome de apnea / hipopnea obstructiva del sueño, narcolepsia y trastornos del sueño en el trabajo por turnos.
En OSAHS, NUVIGIL está indicado como un complemento del tratamiento o tratamientos estándar para la obstrucción subyacente. Si la presión positiva continua en la vía aérea (CPAP) es el tratamiento de elección para un paciente, se debe realizar un esfuerzo máximo para tratar con CPAP durante un período de tiempo adecuado antes de iniciar NUVIGIL. Si NUVIGIL se usa de forma complementaria con CPAP, es necesario alentar y evaluar periódicamente el cumplimiento de CPAP.
En todos los casos, la atención cuidadosa al diagnóstico y tratamiento de los trastornos del sueño subyacentes es de suma importancia. Los prescriptores deben tener en cuenta que algunos pacientes pueden tener más de un trastorno del sueño que contribuye a su somnolencia excesiva.
La efectividad de NUVIGIL en el uso a largo plazo (más de 12 semanas) no se ha evaluado sistemáticamente en ensayos controlados con placebo. El médico que elige recetar NUVIGIL por un tiempo prolongado en los pacientes debe reevaluar periódicamente la utilidad a largo plazo para el paciente individual.
parte superior
Contraindicaciones
NUVIGIL está contraindicado en pacientes con hipersensibilidad conocida al modafinilo y al armodafinilo o sus ingredientes inactivos.
parte superior
Advertencias
Erupción grave, incluido el síndrome de Stevens-Johnson
Se ha informado sarpullido grave que requiere hospitalización e interrupción del tratamiento en adultos en asociación con el uso de armodafinilo y en adultos y niños en asociación con el uso de modafinilo, una mezcla racémica de modafinilo S y R (este último es armodafinilo).
El armodafinilo no se ha estudiado en pacientes pediátricos en ningún entorno y no está aprobado para su uso en pacientes pediátricos por ninguna indicación.
No se han informado erupciones cutáneas graves en ensayos clínicos en adultos (0 por 1,595) de armodafinilo. Sin embargo, se han informado casos de erupción cutánea grave en adultos con experiencia posterior a la comercialización. Debido a que el armodafinilo es el isómero R del modafinilo racémico, no se puede descartar un riesgo similar de erupción cutánea grave en pacientes pediátricos con armodafinilo.
En los ensayos clínicos de modafinilo (el racemato), la incidencia de erupción cutánea que provocó la interrupción fue de aproximadamente el 0,8% (13 por 1.585) en pacientes pediátricos (edad <17 años); Estas erupciones incluyeron 1 caso de posible síndrome de Stevens-Johnson (SJS) y 1 caso de reacción aparente de hipersensibilidad multiorgánica. Varios de los casos se asociaron con fiebre y otras anormalidades (por ejemplo, vómitos, leucopenia). La mediana del tiempo hasta la erupción que resultó en la interrupción fue de 13 días. No se observaron tales casos entre 380 pacientes pediátricos que recibieron placebo. No se han informado erupciones cutáneas graves en ensayos clínicos en adultos (0 por 4,264) de modafinilo. Casos raros de erupción cutánea grave o potencialmente mortal, incluidos SSJ, necrólisis epidérmica tóxica (NET) y erupción farmacológica con Se han reportado eosinofilia y síntomas sistémicos (DRESS) en adultos y niños en la experiencia mundial posterior a la comercialización con modafinilo La tasa de notificación de TEN y SJS asociada con el uso de modafinilo, que generalmente se acepta como una subestimación debido al subregistro, excede la tasa de incidencia de fondo. Las estimaciones de la tasa de incidencia de fondo para estas reacciones cutáneas graves en la población general oscilan entre 1 y 2 casos por millón de personas años.
No se conocen factores que predigan el riesgo de ocurrencia o la gravedad de la erupción asociada con armodafinilo o modafinilo. Casi todos los casos de erupción cutánea grave asociada con armodafinilo o modafinilo ocurrieron dentro de 1 a 5 semanas después del inicio del tratamiento. Sin embargo, se han informado casos aislados después de un tratamiento prolongado con modafinilo (por ejemplo, 3 meses). En consecuencia, no se puede confiar en la duración de la terapia como un medio para predecir el riesgo potencial anunciado por la primera aparición de una erupción.
Aunque las erupciones benignas también ocurren con armodafinilo, no es posible predecir de manera confiable qué erupciones resultarán serias. En consecuencia, el armodafinilo normalmente debe suspenderse al primer signo de erupción, a menos que la erupción claramente no esté relacionada con el medicamento. La interrupción del tratamiento no puede evitar que una erupción se convierta en una amenaza para la vida o en una discapacidad o desfiguración permanente.
Angioedema y reacciones anafilactoides.
Se observó un caso grave de angioedema y un caso de hipersensibilidad (con erupción cutánea, disfagia y broncoespasmo) entre 1,595 pacientes tratados con armodafinilo. Se debe aconsejar a los pacientes que interrumpan la terapia e inmediatamente informen a su médico cualquier signo o síntomas que sugieren angioedema o anafilaxia (por ejemplo, hinchazón de la cara, ojos, labios, lengua o laringe; dificultad para tragar o respirar; ronquera).
Reacciones de hipersensibilidad multiorgánica
Las reacciones de hipersensibilidad multiorgánica, incluida al menos una muerte en la experiencia posterior a la comercialización, tienen ocurrió en estrecha asociación temporal (tiempo medio de detección 13 días: rango 4-33) hasta el inicio de modafinilo No se puede descartar un riesgo similar de reacciones de hipersensibilidad multiorgánica con armodafinilo.
Aunque ha habido un número limitado de informes, las reacciones de hipersensibilidad multiorgánica pueden provocar hospitalización o poner en peligro la vida. No se conocen factores que predigan el riesgo de ocurrencia o la gravedad de las reacciones de hipersensibilidad multiorgánica asociadas con modafinilo. Los signos y síntomas de este trastorno fueron diversos; sin embargo, los pacientes típicamente, aunque no exclusivamente, presentaron fiebre y erupción cutánea asociada con otro compromiso del sistema orgánico. Otras manifestaciones asociadas incluyeron miocarditis, hepatitis, anomalías en las pruebas de función hepática, anomalías hematológicas (p. ej., eosinofilia, leucopenia, trombocitopenia), prurito y astenia. Debido a que la hipersensibilidad multiorgánica es variable en su expresión, pueden aparecer otros síntomas y signos del sistema orgánico, que no se mencionan aquí.
Si se sospecha una reacción de hipersensibilidad multiorgánica, se debe suspender NUVIGIL. Aunque no hay informes de casos que indiquen sensibilidad cruzada con otras drogas que producen este síndrome, la experiencia con medicamentos asociados con hipersensibilidad multiorgánica indicaría que esto es un posibilidad.
Somnolencia persistente
Se debe informar a los pacientes con niveles anormales de somnolencia que toman NUVIGIL que su nivel de vigilia puede no volver a la normalidad. Los pacientes con somnolencia excesiva, incluidos los que toman NUVIGIL, deben ser reevaluados con frecuencia por sus grado de somnolencia y, si corresponde, se recomienda evitar conducir o cualquier otro peligro potencial actividad. Los prescriptores también deben saber que los pacientes pueden no reconocer la somnolencia o la somnolencia hasta que se les pregunte directamente sobre la somnolencia o la somnolencia durante actividades específicas.
Síntomas psiquiátricos
Se han informado experiencias adversas psiquiátricas en pacientes tratados con modafinilo. El modafinilo y el armodafinilo (NUVIGIL) están muy relacionados. Por lo tanto, se espera que la incidencia y el tipo de síntomas psiquiátricos asociados con el armodafinilo sean similares a la incidencia y el tipo de estos eventos con el modafinilo.
Los eventos adversos posteriores a la comercialización asociados con el uso de modafinilo han incluido manía, delirios, alucinaciones, ideación suicida y agresión, algunos de los cuales resultaron en hospitalización. Muchos, pero no todos, los pacientes tenían antecedentes psiquiátricos previos. Un voluntario masculino sano desarrolló ideas de referencia, delirios paranoides y alucinaciones auditivas en asociación con múltiples dosis diarias de 600 mg de modafinilo y privación del sueño. No hubo evidencia de psicosis 36 horas después de la interrupción del fármaco.
En el ensayo controlado de la base de datos NUVIGIL, la ansiedad, la agitación, el nerviosismo y la irritabilidad fueron razones para interrupción del tratamiento con mayor frecuencia en pacientes con NUVIGIL en comparación con placebo (NUVIGIL 1.2% y placebo 0.3%). En los estudios controlados por NUVIGIL, la depresión también fue una razón para la interrupción del tratamiento con mayor frecuencia en pacientes con NUVIGIL en comparación con placebo (NUVIGIL 0.6% y placebo 0.2%). Se observaron dos casos de ideación suicida en ensayos clínicos. Se debe tener precaución cuando se administra NUVIGIL a pacientes con antecedentes de psicosis, depresión o manía. Si se desarrollan síntomas psiquiátricos en asociación con la administración de NUVIGIL, considere suspender NUVIGIL.
parte superior
PRECAUCIONES
Diagnóstico de los trastornos del sueño.
NUVIGIL debe usarse solo en pacientes que hayan tenido una evaluación completa de su somnolencia excesiva y en quienes El diagnóstico de narcolepsia, SAHS y / o SWSD se ha realizado de acuerdo con los criterios de diagnóstico de ICSD o DSM (Ver Ensayos clínicos). Dicha evaluación generalmente consiste en una historia clínica completa y un examen físico, y puede complementarse con pruebas en un entorno de laboratorio. Algunos pacientes pueden tener más de un trastorno del sueño que contribuye a su somnolencia excesiva (por ejemplo, OSAHS y SWSD coincidentes en el mismo paciente).
Uso de CPAP en pacientes con SAHS
En OSAHS, NUVIGIL está indicado como un complemento del tratamiento o tratamientos estándar para la obstrucción subyacente. Si la presión positiva continua en la vía aérea (CPAP) es el tratamiento de elección para un paciente, se debe realizar un esfuerzo máximo para tratar con CPAP durante un período de tiempo adecuado antes de iniciar NUVIGIL. Si NUVIGIL se usa de forma complementaria con CPAP, es necesario alentar y evaluar periódicamente el cumplimiento de CPAP. Hubo una ligera tendencia a reducir el uso de CPAP a lo largo del tiempo (reducción media de 18 minutos para pacientes tratados con NUVIGIL y una reducción de 6 minutos para los pacientes tratados con placebo de un uso promedio inicial de 6.9 horas por noche) en NUVIGIL juicios.
General
Aunque no se ha demostrado que NUVIGIL produzca un deterioro funcional, cualquier medicamento que afecte al SNC puede alterar el juicio, el pensamiento o las habilidades motoras. Se debe advertir a los pacientes sobre la operación de un automóvil u otra maquinaria peligrosa hasta que estén razonablemente seguro de que la terapia con NUVIGIL no afectará negativamente su capacidad para participar en tales ocupaciones.
Sistema cardiovascular
NUVIGIL no se ha evaluado ni utilizado de manera apreciable en pacientes con antecedentes recientes de infarto de miocardio o angina inestable, y dichos pacientes deben ser tratados con precaución.
En estudios clínicos de PROVIGIL, los signos y síntomas incluyen dolor torácico, palpitaciones, disnea e isquemia transitoria. Se observaron cambios en la onda T en el ECG en tres sujetos en asociación con prolapso de la válvula mitral o ventricular izquierdo hipertrofia. Se recomienda que los comprimidos de NUVIGIL no se utilicen en pacientes con antecedentes de hipertrofia ventricular izquierda o en pacientes con prolapso de la válvula mitral que han experimentado el síndrome de prolapso de la válvula mitral cuando recibieron SNC previamente estimulantes Los signos del síndrome de prolapso de la válvula mitral incluyen, entre otros, cambios isquémicos en el ECG, dolor torácico o arritmia. Si se produce un nuevo inicio de cualquiera de estos síntomas, considere la evaluación cardíaca.
La monitorización de la presión arterial en ensayos controlados a corto plazo (‰ ¤3 meses) mostró solo pequeños aumentos promedio en la media sistólica y presión arterial diastólica en pacientes que reciben NUVIGIL en comparación con placebo (1,2 a 4,3 mmHg en los diversos experimentos grupos). También hubo una proporción ligeramente mayor de pacientes en NUVIGIL que requirieron un uso nuevo o mayor de medicamentos antihipertensivos (2.9%) en comparación con los pacientes que recibieron placebo (1.8%). El aumento de la monitorización de la presión arterial puede ser apropiado en pacientes con NUVIGIL.
Pacientes que usan anticonceptivos esteroideos
La efectividad de los anticonceptivos esteroides puede reducirse cuando se usa con NUVIGIL y durante un mes después de la interrupción de la terapia (ver Precauciones, Interacciones con la drogas). Se recomiendan métodos anticonceptivos alternativos o concomitantes para pacientes tratadas con NUVIGIL y durante un mes después de la interrupción del tratamiento con NUVIGIL.
Pacientes que usan ciclosporina
Los niveles sanguíneos de ciclosporina pueden reducirse cuando se usan con NUVIGIL (Ver Precauciones, Interacciones con la drogas). El monitoreo de las concentraciones circulantes de ciclosporina y el ajuste de dosis apropiado para la ciclosporina deben considerarse cuando estos medicamentos se usan concomitantemente.
Pacientes con insuficiencia hepática severa
En pacientes con insuficiencia hepática grave, con o sin cirrosis (ver Farmacología Clínica), NUVIGIL debe administrarse a una dosis reducida (ver Dosificación y administración).
Pacientes con insuficiencia renal severa
No hay información adecuada para determinar la seguridad y la eficacia de la dosificación en pacientes con insuficiencia renal grave (Para farmacocinética en insuficiencia renal, ver Farmacología Clínica).
Pacientes de edad avanzada
En pacientes de edad avanzada, la eliminación de armodafinilo y sus metabolitos puede reducirse como consecuencia del envejecimiento. Por lo tanto, se debe considerar el uso de dosis más bajas en esta población (Ver Farmacología Clínica y Dosificación y administración).
Información para pacientes
Se aconseja a los médicos que discutan los siguientes problemas con los pacientes a quienes les recetan NUVIGIL.
NUVIGIL está indicado para pacientes con niveles anormales de somnolencia. Se ha demostrado que NUVIGIL mejora, pero no elimina, esta tendencia anormal a quedarse dormido. Por lo tanto, los pacientes no deben alterar su comportamiento anterior con respecto a actividades potencialmente peligrosas (por ejemplo, conducir, operar maquinaria) u otros actividades que requieren niveles apropiados de vigilia, hasta que se haya demostrado que el tratamiento con NUVIGIL produce niveles de vigilia que permiten tal ocupaciones. Se debe informar a los pacientes que NUVIGIL no reemplaza el sueño.
Se debe informar a los pacientes que puede ser crítico que continúen tomando sus tratamientos previamente recetados (por ejemplo, los pacientes con SAHS que reciben CPAP deben continuar haciéndolo).
Se debe informar a los pacientes sobre la disponibilidad de un folleto de información para el paciente, y se les debe indicar que lo lean antes de tomar NUVIGIL. Consulte la Información del paciente al final de este etiquetado para ver el texto del folleto proporcionado a los pacientes.
Se debe recomendar a los pacientes que se pongan en contacto con su médico si experimentan erupción cutánea, depresión, ansiedad o signos de psicosis o manía.
El embarazo
Se debe aconsejar a las pacientes que notifiquen a su médico si quedan embarazadas o si tienen la intención de quedar embarazadas durante la terapia. Se debe advertir a las pacientes sobre el posible aumento del riesgo de embarazo al usar anticonceptivos esteroideos (incluidos los de depósito o implantables). anticonceptivos) con NUVIGIL y durante un mes después de la interrupción de la terapia (ver carcinogénesis, mutagénesis, deterioro de la fertilidad y El embarazo).
Enfermería
Se debe aconsejar a los pacientes que notifiquen a su médico si están amamantando a un bebé.
Medicación concomitante
Se debe aconsejar a los pacientes que informen a su médico si están tomando, o planean tomar, cualquier medicamentos recetados o de venta libre, debido al potencial de interacciones entre NUVIGIL y otras drogas.
Alcohol
Se debe informar a los pacientes que no se ha estudiado el uso de NUVIGIL en combinación con alcohol. Se debe advertir a los pacientes que es prudente evitar el alcohol mientras toman NUVIGIL.
Reacciones alérgicas
Se debe recomendar a los pacientes que dejen de tomar NUVIGIL y que notifiquen a su médico si desarrollan una erupción cutánea. urticaria, llagas en la boca, ampollas, descamación de la piel, dificultad para tragar o respirar o una alergia relacionada fenómeno.
Interacciones con la drogas
Posibles interacciones con medicamentos que inhiben, inducen o se metabolizan por las isoenzimas del citocromo P450 y otras enzimas hepáticas
Debido a la participación parcial de las enzimas CYP3A en la eliminación metabólica de armodafinilo, la administración conjunta de inductores potentes de CYP3A4 / 5 (p. ej., carbamazepina, fenobarbital, rifampicina) o inhibidores de CYP3A4 / 5 (p. ej., ketoconazol, eritromicina) podrían alterar los niveles plasmáticos de armodafinilo
El potencial de NUVIGIL para alterar el metabolismo de otras drogas por inducción o inhibición enzimática
Medicamentos metabolizados por CYP1A2: Los datos in vitro demostraron que el armodafinilo muestra una respuesta inductiva débil para CYP1A2 y posiblemente CYP3A actividades de una manera relacionada con la concentración y demostró que la actividad CYP2C19 es reversiblemente inhibida por armodafinilo Sin embargo, el efecto sobre la actividad de CYP1A2 no se observó clínicamente en un estudio de interacción realizado con cafeína (ver Farmacología Clínica, Farmacocinética, interacciones farmacológicas).
Medicamentos metabolizados por CYP3A4 / 5 (por ejemplo, ciclosporina, etinilestradiol, midazolam y triazolam): La administración crónica de NUVIGIL resultó en una inducción moderada de la actividad de CYP3A. Por lo tanto, la efectividad de los medicamentos que son sustratos para las enzimas CYP3A (por ejemplo, ciclosporina, etinilo estradiol, midazolam y triazolam) pueden reducirse después del inicio del tratamiento concurrente con NUVIGIL. Se observó una reducción del 32% en la exposición sistémica de midazolam oral tras la administración concomitante de armodafinilo con midazolam. Puede ser necesario ajustar la dosis (ver Farmacología Clínica, Farmacocinética, interacciones farmacológicas). Tales efectos (concentraciones reducidas) también se observaron con la administración concomitante de modafinilo con ciclosporina, etinilestradiol y triazolam.
Medicamentos metabolizados por CYP2C19 (por ejemplo, omeprazol, diazepam, fenitoína y propranolol): La administración de NUVIGIL resultó en una inhibición moderada de la actividad de CYP2C19. Por lo tanto, la reducción de la dosis puede ser necesaria para algunos medicamentos que son sustratos del CYP2C19 (por ejemplo, fenitoína, diazepam y propranolol, omeprazol y clomipramina) cuando se usan simultáneamente con NUVIGIL. Se observó un aumento del 40% en la exposición tras la administración concomitante de armodafinilo con omeprazol. (Ver Farmacología Clínica, Farmacocinética, interacciones farmacológicas).
Interacciones con el SNC ADrogas activas
No se dispone de datos específicos sobre el potencial de interacción fármaco-fármaco del armodafinilo con fármacos activos del SNC. Sin embargo, la siguiente información disponible de interacción farmacológica sobre modafinilo debería ser aplicable al armodafinilo (Ver Descripción y Farmacología Clínica).
La administración concomitante de modafinilo con metilfenidato o dextroanfetamina no produjo alteraciones significativas en el perfil farmacocinético de modafinilo o cualquier estimulante, aunque la absorción de modafinilo se retrasó aproximadamente uno hora.
El modafinilo o la clomipramina concomitantes no alteraron el perfil PK de ninguno de los fármacos; sin embargo, se informó un incidente de aumento de los niveles de clomipramina y su metabolito activo desmetilclomipramina en un paciente con narcolepsia durante el tratamiento con modafinilo.
No se dispone de datos específicos sobre el potencial de interacción fármaco-fármaco de armodafinilo o modafinilo con inhibidores de la monoaminooxidasa (MAO). Por lo tanto, se debe tener precaución al administrar concomitantemente inhibidores de la MAO y NUVIGIL.
Interacciones con otras drogas
No se dispone de datos específicos sobre el potencial de interacción fármaco-fármaco del armodafinilo para otros fármacos adicionales. Sin embargo, la siguiente información disponible de interacción farmacológica sobre modafinilo debería ser aplicable al armodafinilo.
Warfarina - La administración concomitante de modafinilo con warfarina no produjo cambios significativos en los perfiles farmacocinéticos de warfarina R y S. Sin embargo, dado que solo se probó una dosis única de warfarina en este estudio, no se puede descartar una interacción farmacodinámica. Por lo tanto, se debe considerar un monitoreo más frecuente de los tiempos de protrombina / INR cada vez que NUVIGIL se coadministra con warfarina.
Carcinogénesis, Mutagénesis, Deterioro de la Fertilidad
Carcinogénesis
No se han realizado estudios de carcinogenicidad con armodafinilo solo. Se realizaron estudios de carcinogenicidad en los que se administró modafinilo en la dieta a ratones durante 78 semanas y a ratas durante 104 semanas a dosis de 6, 30 y 60 mg / kg / día. La dosis más alta estudiada representa 1.5 (ratón) o 3 (rata) veces mayor que la dosis diaria recomendada para adultos de modafinilo (200 mg) en una base de mg / m2. No hubo evidencia de tumorigénesis asociada con la administración de modafinilo en estos estudios. Sin embargo, dado que el estudio en ratones utilizó una dosis alta inadecuada que no era representativa de una dosis máxima tolerada, se realizó un estudio de carcinogenicidad posterior en la Tg. Ratón AC transgénico. Dosis evaluadas en la Tg. El ensayo de CA fue de 125, 250 y 500 mg / kg / día, administrados por vía cutánea. No hubo evidencia de tumorigenicidad asociada con la administración de modafinilo; sin embargo, este modelo dérmico puede no evaluar adecuadamente el potencial carcinogénico de un medicamento administrado por vía oral.
Mutagénesis
El armodafinilo se evaluó en un ensayo de mutación inversa bacteriana in vitro y en un ensayo de aberración cromosómica de mamíferos in vitro en linfocitos humanos. El armodafinilo fue negativo en estos ensayos, tanto en ausencia como en presencia de activación metabólica.
El modafinilo no demostró evidencia de potencial mutagénico o clastogénico en una serie de ensayos in vitro (es decir, ensayo de mutación inversa bacteriana, ensayo de linfoma de ratón tk, aberración cromosómica ensayo en linfocitos humanos, ensayo de transformación celular en células embrionarias de ratón BALB / 3T3) ensayos en ausencia o presencia de activación metabólica, o in vivo (micronúcleo de médula ósea de ratón) ensayos El modafinilo también fue negativo en el ensayo de síntesis de ADN no programado en hepatocitos de rata.
Deterioro de la fertilidad
No se realizó un estudio de fertilidad y desarrollo embrionario temprano (hasta la implantación) con armodafinilo solo.
Administración oral de modafinilo (dosis de hasta 480 mg / kg / día) a ratas machos y hembras antes y durante el apareamiento y continuar en las hembras hasta el día 7 de gestación produjo un aumento en el tiempo de apareamiento en el momento más alto dosis; No se observaron efectos sobre otros parámetros de fertilidad o reproducción. La dosis sin efecto de 240 mg / kg / día se asoció con una exposición plasmática de modafinilo (AUC) aproximadamente igual a la de los humanos a la dosis recomendada de 200 mg.
El embarazo
Embarazo Categoría C.
En estudios realizados en ratas (armodafinilo, modafinilo) y conejos (modafinilo), se observó toxicidad en el desarrollo en exposiciones clínicamente relevantes.
La administración oral de armodafinilo (60, 200 o 600 mg / kg / día) a ratas preñadas durante todo el período de organogénesis resultó en un aumento incidencias de las variaciones viscerales y esqueléticas fetales en la dosis intermedia o mayor y disminución de los pesos corporales fetales en la dosis más alta dosis. La dosis sin efecto para la toxicidad del desarrollo embriofetal de rata se asoció con un armodafinilo en plasma. exposición (AUC) aproximadamente 0.03 veces el AUC en humanos a la dosis diaria máxima recomendada de 250 mg.
Modafinilo (50, 100 o 200 mg / kg / día) administrado por vía oral a ratas preñadas durante todo el período de organogénesis causada, en ausencia de toxicidad materna, un aumento en las reabsorciones y una mayor incidencia de variaciones viscerales y esqueléticas en la descendencia en el nivel más alto dosis. La dosis más alta sin efecto para la toxicidad del desarrollo embriofetal de rata se asoció con un plasma exposición a modafinilo aproximadamente 0.5 veces el AUC en humanos a la dosis diaria recomendada (RHD) de 200 mg. Sin embargo, en un estudio posterior de hasta 480 mg / kg / día (exposición de modafinilo en plasma aproximadamente 2 veces el AUC en humanos en el RHD) no se observaron efectos adversos sobre el desarrollo embriofetal.
Modafinilo administrado por vía oral a conejos preñadas durante todo el período de organogénesis a dosis de hasta 100 mg / kg / día (el AUC de modafinilo en plasma aproximadamente igual al AUC en humanos en el RHD) no tuvo efecto sobre el embrio-fetal desarrollo; sin embargo, las dosis utilizadas fueron demasiado bajas para evaluar adecuadamente los efectos del modafinilo en el desarrollo embriofetal. En un estudio posterior de toxicidad del desarrollo que evalúa dosis de 45, 90 y 180 mg / kg / día en embarazadas conejos, las incidencias de alteraciones estructurales fetales y muerte embriofetal se incrementaron al máximo dosis. La dosis más alta sin efecto para la toxicidad del desarrollo se asoció con un AUC de modafinilo en plasma aproximadamente igual al AUC en humanos en el RHD.
La administración de modafinilo a ratas durante la gestación y la lactancia a dosis orales de hasta 200 mg / kg / día resultó en una disminución viabilidad en la descendencia a dosis mayores de 20 mg / kg / día (AUC modafinilo en plasma aproximadamente 0.1 veces el AUC en humanos en el RHD). No se observaron efectos sobre el desarrollo postnatal y los parámetros neuroconductuales en las crías supervivientes.
No existen estudios adecuados y bien controlados de armodafinilo o modafinilo en mujeres embarazadas. Se han notificado dos casos de retraso del crecimiento intrauterino y un caso de aborto espontáneo en asociación con armodafinilo y modafinilo. Aunque la farmacología del armodafinilo no es idéntica a la de las aminas simpaticomiméticas, sí comparte algunas propiedades farmacológicas con esta clase. Algunos de estos medicamentos se han asociado con retraso del crecimiento intrauterino y abortos espontáneos. Se desconoce si los casos informados con armodafinilo están relacionados con las drogas.
El armodafinilo o el modafinilo deben usarse durante el embarazo solo si el beneficio potencial justifica el riesgo potencial para el feto.
Trabajo y entrega
El efecto del armodafinilo sobre el trabajo de parto y el parto en humanos no se ha investigado sistemáticamente.
Madres lactantes
No se sabe si el armodafinilo o sus metabolitos se excretan en la leche humana. Debido a que muchos medicamentos se excretan en la leche humana, se debe tener precaución cuando se administran tabletas NUVIGIL a una mujer lactante.
Uso pediátrico
No se ha establecido la seguridad y eficacia del uso de armodafinilo en personas menores de 17 años. Se ha observado sarpullido grave en pacientes pediátricos que reciben modafinilo.
Uso geriátrico
No se ha establecido la seguridad y eficacia en personas mayores de 65 años.
parte superior
Reacciones adversas
Se ha evaluado la seguridad del armodafinilo en más de 1100 pacientes con somnolencia excesiva asociada con trastornos primarios del sueño y la vigilia. En ensayos clínicos, se ha encontrado que NUVIGIL es generalmente bien tolerado y la mayoría de las experiencias adversas fueron leves a moderadas.
En los estudios clínicos controlados con placebo, los eventos adversos más comúnmente observados (â ‰ ¥ 5%) asociados con el uso de NUVIGIL que ocurre con más frecuencia que en los pacientes tratados con placebo fueron dolor de cabeza, náuseas, mareos y insomnio. El perfil de eventos adversos fue similar en todos los estudios.
En los ensayos clínicos controlados con placebo, 44 de los 645 pacientes (7%) que recibieron NUVIGIL interrumpieron debido a una experiencia adversa en comparación con 16 de los 445 (4%) de los pacientes que recibieron placebo. La razón más frecuente para la interrupción fue dolor de cabeza (1%).
Incidencia en ensayos controlados
La siguiente tabla (Tabla 3) presenta las experiencias adversas que ocurrieron a una tasa de 1% o más y fueron más frecuente en pacientes tratados con NUVIGIL que en pacientes del grupo placebo en los ensayos clínicos controlados con placebo.
El prescriptor debe ser consciente de que las cifras proporcionadas a continuación no pueden usarse para predecir la frecuencia de las experiencias adversas en el curso de la práctica médica habitual, donde las características del paciente y otros factores pueden diferir de los que ocurren durante la clínica estudios. Del mismo modo, las frecuencias citadas no se pueden comparar directamente con las cifras obtenidas de otras investigaciones clínicas que involucran diferentes tratamientos, usos o investigadores. Sin embargo, la revisión de estas frecuencias proporciona a los prescriptores una base para estimar la contribución relativa de los factores farmacológicos y no farmacológicos a la incidencia de eventos adversos en la población estudiada.
| Sistema de clasificación de órganos Término preferido de MedDRA |
Nuvigil (Porcentaje, N = 645) |
Placebo (Porcentaje, N = 445) |
ª Cuatro estudios clínicos doble ciego, controlados con placebo en SWSD, OSAHS y narcolepsia; la incidencia se redondea al porcentaje entero más cercano. Se incluyen solo aquellos eventos para los cuales la incidencia de Nuvigil es mayor que la del placebo. | ||
| Trastornos cardíacos | ||
| Palpitaciones | 2 | 1 |
| Desórdenes gastrointestinales | ||
| Náusea | 7 | 3 |
| Diarrea | 4 | 2 |
| Boca seca | 4 | 1 |
| Dispepsia | 2 | 0 |
| Dolor abdominal superior | 2 | 1 |
| Estreñimiento | 1 | 0 |
| Vómitos | 1 | 0 |
| Heces sueltas | 1 | 0 |
| Desordenes generales y condiciones administrativas del sitio | ||
| Fatiga | 2 | 1 |
| Sed | 1 | 0 |
| Enfermedad similar a la influenza | 1 | 0 |
| Dolor | 1 | 0 |
| Pirexia | 1 | 0 |
| Trastornos del sistema inmunitario | ||
| Alergia estacional | 1 | 0 |
| Investigaciones | ||
| Aumento de la gamma-glutamiltransferasa | 1 | 0 |
| Aumento de la frecuencia cardíaca | 1 | 0 |
| Trastornos del metabolismo y la nutrición. | ||
| Anorexia | 1 | 0 |
| Disminucion del apetito | 1 | 0 |
| Trastornos del sistema nervioso | ||
| Dolor de cabeza | 17 | 9 |
| Mareo | 5 | 2 |
| Perturbación en la atención | 1 | 0 |
| Temblor | 1 | 0 |
| Migraña | 1 | 0 |
| Paraesthia | 1 | 0 |
| Desórdenes psiquiátricos | ||
| Insomnio | 5 | 1 |
| Ansiedad | 4 | 1 |
| Depresión | 2 | 0 |
| Agitación | 1 | 0 |
| Nerviosismo | 1 | 0 |
| Estado de ánimo deprimido | 1 | 0 |
| Trastornos renales y urinarios | ||
| Poliuria | 1 | 0 |
| Trastornos respiratorios, torácicos y mediastínicos. | ||
| Disnea | 1 | 0 |
| Trastornos de la piel y del tejido subcutáneo | ||
| Erupción | 2 | 0 |
| Dermatitis de contacto | 1 | 0 |
| Hiperhidrosis | 1 | 0 |
Dependencia de dosis de eventos adversos
En los ensayos clínicos controlados con placebo que compararon dosis de 150 mg / día y 250 mg / día de Nuvigil y placebo, Los únicos eventos adversos que parecían estar relacionados con la dosis fueron dolor de cabeza, erupción cutánea, depresión, boca seca, insomnio y náusea.
ª Cuatro estudios clínicos doble ciego controlados con placebo en SWSD, OSAHS y narcolepsia. | ||||
| Sistema de clasificación de órganos Término preferido de MedDRA |
Nuvigil 250 mg (Por ciento, N = 198) |
Nuvigil 150 mg (Por ciento, N = 447) |
Nuvigil Conjunto (Por ciento, N = 645) |
Placebo (Por ciento, N = 445) |
|---|---|---|---|---|
| Desórdenes gastrointestinales | ||||
| Náusea | 9 | 6 | 7 | 3 |
| Boca seca | 7 | 2 | 4 | <1 |
| Trastornos del sistema nervioso | ||||
| Dolor de cabeza | 23 | 14 | 17 | 9 |
| Desórdenes psiquiátricos | ||||
| Insomnio | 6 | 4 | 5 | 1 |
| Depresión | 3 | 1 | 2 | <1 |
| Trastornos de la piel y del tejido subcutáneo | ||||
| Erupción | 4 | 1 | 2 | <1 |
Cambios de signos vitales
Hubo aumentos pequeños, pero consistentes, en los valores promedio de la presión arterial sistólica y diastólica media en ensayos controlados (ver Precauciones). Hubo un pequeño, pero constante, aumento promedio en la frecuencia del pulso sobre el placebo en ensayos controlados. Este aumento varió de 0.9 a 3.5 BPM.
Cambios de laboratorio
Los parámetros de química clínica, hematología y análisis de orina se monitorearon en los estudios. Los niveles plasmáticos medios de gamma glutamiltransferasa (GGT) y fosfatasa alcalina (AP) fueron mayores después de la administración de NUVIGIL, pero no placebo. Pocos sujetos, sin embargo, tuvieron elevaciones de GGT o AP fuera del rango normal. No se observaron diferencias en alanina aminotransferasa, aspartato aminotransferasa, proteína total, albúmina, o bilirrubina total, aunque hubo casos raros de elevaciones aisladas de AST y / o ALT. Se observó un solo caso de pancitopenia leve después de 35 días de tratamiento y se resolvió con la interrupción del fármaco. Se observó una pequeña disminución media desde el inicio en el ácido úrico sérico en comparación con el placebo en los ensayos clínicos. La importancia clínica de este hallazgo es desconocida.
Cambios de ECG
No se pudo atribuir ningún patrón de anormalidades del ECG a la administración de NUVIGIL en ensayos clínicos controlados con placebo.
parte superior
Abuso y dependencia de drogas
Clase de sustancia controlada
El armodafinilo (NUVIGIL) es una sustancia controlada de la Lista IV.
Potencial de abuso y dependencia
Aunque el potencial de abuso del armodafinilo no se ha estudiado específicamente, es probable que su potencial de abuso sea similar al del modafinilo (PROVIGIL). En humanos, el modafinilo produce efectos psicoactivos y eufóricos, alteraciones en el estado de ánimo, la percepción, el pensamiento y los sentimientos típicos de otros estimulantes del SNC. En estudios de unión in vitro, el modafinilo se une al sitio de recaptación de dopamina y provoca un aumento en la dopamina extracelular, pero no aumenta la liberación de dopamina. El modafinilo se está reforzando, como lo demuestra su autoadministración en monos previamente entrenados para autoadministrarse cocaína. En algunos estudios, el modafinilo también fue parcialmente discriminado como estimulante. Los médicos deben seguir de cerca a los pacientes, especialmente aquellos con antecedentes de abuso de drogas y / o estimulantes (por ejemplo, metilfenidato, anfetaminas o cocaína). Se debe observar a los pacientes en busca de signos de mal uso o abuso (por ejemplo, aumento de dosis o comportamiento de búsqueda de drogas).
El potencial de abuso de modafinilo (200, 400 y 800 mg) se evaluó en relación con el metilfenidato (45 y 90 mg) en un estudio de pacientes hospitalizados en individuos con drogas de abuso. Los resultados de este estudio clínico demostraron que el modafinilo produce efectos psicoactivos y eufóricos y sensaciones consistentes con otros estimulantes programados del SNC (metilfenidato).
parte superior
Sobredosis
Experiencia humana
No se informaron sobredosis en los estudios clínicos de NUVIGIL. Es probable que los síntomas de sobredosis de NUVIGIL sean similares a los del modafinilo. La sobredosis en los ensayos clínicos de modafinilo incluyó excitación o agitación, insomnio y elevaciones leves o moderadas en los parámetros hemodinámicos. Desde la experiencia posterior a la comercialización con modafinilo, no ha habido informes de sobredosis fatales que involucren modafinilo solo (dosis de hasta 12 gramos). Las sobredosis que involucran múltiples medicamentos, incluido el modafinilo, han tenido resultados fatales. Se han incluido los síntomas que con mayor frecuencia acompañan a una sobredosis de modafinilo, solo o en combinación con otros medicamentos; insomnio; síntomas del sistema nervioso central tales como inquietud, desorientación, confusión, excitación y alucinaciones; cambios digestivos como náuseas y diarrea; y cambios cardiovasculares como taquicardia, bradicardia, hipertensión y dolor torácico.
Manejo de sobredosis
No existe un antídoto específico para los efectos tóxicos de una sobredosis de NUVIGIL. Tales sobredosis deben manejarse con atención principalmente de apoyo, incluida la monitorización cardiovascular. Si no hay contraindicaciones, se debe considerar la emesis inducida o el lavado gástrico. No hay datos que sugieran la utilidad de la diálisis o la acidificación o alcalinización urinaria para mejorar la eliminación del fármaco. El médico debe considerar ponerse en contacto con un centro de control de intoxicaciones para recibir asesoramiento sobre el tratamiento de cualquier sobredosis.
parte superior
Dosificación y administración
Síndrome de apnea obstructiva del sueño / hipopnea (SAHS) y narcolepsia
La dosis recomendada de NUVIGIL para pacientes con SAHS o narcolepsia es de 150 mg o 250 mg administrados como una dosis única en la mañana. En pacientes con SAHS, las dosis de hasta 250 mg / día, administradas como dosis única, han sido bien toleradas, pero existe No hay evidencia consistente de que esta dosis confiera un beneficio adicional más allá de la dosis de 150 mg / día (Ver Farmacología Clínica y Ensayos clínicos).
Trastorno del sueño por turnos de trabajo (SWSD)
La dosis recomendada de NUVIGIL para pacientes con SWSD es de 150 mg administrados diariamente aproximadamente 1 hora antes del inicio de su turno de trabajo.
Se debe considerar el ajuste de la dosis para los medicamentos concomitantes que son sustratos del CYP3A4 / 5, como los anticonceptivos esteroideos, el triazolam y la ciclosporina (ver PRECAUCIONES, Interacciones farmacológicas).
Los medicamentos que se eliminan en gran medida a través del metabolismo CYP2C19, como el diazepam, el propranolol y la fenitoína pueden tener eliminación prolongada tras la administración conjunta con NUVIGIL y puede requerir reducción de dosis y monitoreo de toxicidad (Ver Precauciones, Interacciones con la drogas).
En pacientes con insuficiencia hepática grave, NUVIGIL debe administrarse a una dosis reducida (ver Farmacología Clínica y Precauciones).
No hay información adecuada para determinar la seguridad y la eficacia de la dosificación en pacientes con insuficiencia renal grave (ver Farmacología Clínica y Precauciones).
En pacientes de edad avanzada, la eliminación de armodafinilo y sus metabolitos puede reducirse como consecuencia del envejecimiento. Por lo tanto, se debe considerar el uso de dosis más bajas en esta población (Ver Farmacología Clínica y Precauciones).
parte superior
Cómo se suministra / Almacenamiento y manipulación
Nuvigil® (armodafinilo) tabletas [C-IV]
50 mg: cada tableta redonda, de color blanco a blanquecino, está grabada con  por un lado y "205" por el otro.
por un lado y "205" por el otro.
NDC 63459-205-60 - Botellas de 60
150 mg: cada tableta ovalada de color blanco a blanqueado se graba con por un lado y "215" por el otro.
NDC 63459-215-60 - Botellas de 60
250 mg: cada tableta ovalada, de color blanco a blanquecino, está grabada con por un lado y "225" por el otro.
NDC 63459-225-60 - Botellas de 60
Almacene a 20 ° - 25 ° C (68 ° - 77 ° F).
Fabricado para:
Cephalon, Inc.
Frazer, PA 19355
última actualización 02/2010
Nuvigil hoja de información para el paciente (en inglés simple)
Información detallada sobre Signos, síntomas, causas, tratamientos de los trastornos del sueño
La información en esta monografía no pretende cubrir todos los usos, instrucciones, precauciones, interacciones de drogas o efectos adversos posibles. Esta información es generalizada y no pretende ser un consejo médico específico. Si tiene preguntas sobre los medicamentos que está tomando o desea obtener más información, consulte con su médico, farmacéutico o enfermero.
de regreso:
~ todos los artículos sobre trastornos del sueño


